 Zaloguj
Zaloguj




Niejednemu elektronikowi normy branżowe kojarzą się wyłącznie negatywnie. Wielostronicowe, dość suche w swojej formie dokumenty wydają się przykrym obowiązkiem i źródłem dodatkowych, zdawałoby się niepotrzebnych, kosztów. Tymczasem doświadczeni producenci i projektanci doskonale zdają sobie sprawę z faktu, że opracowania (zarówno międzynarodowych, jak i lokalnych) komitetów normalizacyjnych są w istocie… niemałą pomocą dla osób zaangażowanych merytorycznie w tworzenie produktu.
W tym artykule posłużymy się aparaturą medyczną jako przykładem działu techniki, który szczególnie mocno opiera się na wymaganiach norm. Wybór ten jest nieprzypadkowy, gdyż niewłaściwie zaprojektowane wyroby medyczne mogą stanowić śmiertelne ryzyko dla pacjentów, a często także dla samego personelu jednostek ochrony zdrowia czy nawet… osób postronnych. Jaskrawość tego przykładu pozwoli nam właściwie zobrazować najważniejsze (z punktu widzenia projektanta hardware’u) aspekty, związane ze zdrowym podejściem do normalizacji. Intencjonalnie pominiemy tutaj obszerne wywody prawne, z jakimi zwykle mają kontakt osoby stykające się w swojej pracy zawodowej z tematyką certyfikacji. Omówimy jedynie najważniejsze pojęcia, które pozwolą lepiej zrozumieć istotę procesów oceny zgodności, wdrażania produktu na rynek, a nade wszystko „projektowania zorientowanego na certyfikację”. Skupimy się przy tym na rynku europejskim, choć ogólne zasady dają się także zastosować do procesów przedwdrożeniowych wymaganych na innych kontynentach.
Najważniejsze pojęcia
Zdecydowana większość wyrobów elektronicznych (pomijając nieliczne przypadki) wymaga, w celu zgodnego z aktualnymi przepisami wdrożenia na rynek, oznakowania produktu znakiem CE (Conformité Européenne). Naniesienie (trwałe) znaku CE na dany wyrób oznacza, że jego producent (lub dystrybutor, który przejmuje na siebie obowiązki producenta – np. w przypadku produktów określanych jako white label) deklaruje, że wyrób spełnia wymagania tzw. dyrektyw nowego podejścia.
Celem zastosowania dyrektyw jest zapewnienie niezbędnego poziomu bezpieczeństwa i to zarówno życia i zdrowia osób (użytkowników, operatorów, pacjentów czy też osób postronnych), jak też środowiska naturalnego czy nawet… infrastruktury technicznej. Przykładem tej ostatniej może być dyrektywa EMC, określająca nie tylko podatność urządzeń na zakłócenia zewnętrzne, ale także dopuszczalne poziomy emisji zaburzeń elektromagnetycznych, które mogłyby zakłócać pracę innych produktów (nie wspominając rzecz jasna o kwestiach narażenia istot żywych na zbyt silne promieniowanie elektromagnetyczne).
Najważniejszymi aktami prawnymi wpływającymi (w sposób pośredni) na pracę zarówno projektantów, jak i producentów urządzeń elektronicznych, są dyrektywy i rozporządzenia unijne. O ile rozporządzenie jest aktem wiążącym, obowiązującym na całym obszarze Unii Europejskiej, o tyle dyrektywa określa cel legislacyjny, który osiągnąć muszą wszystkie państwa UE poprzez wprowadzenie lokalnych aktów prawnych. W codziennej praktyce projektanta elektronika najczęściej niezbędne są następujące akty:
- 2014/35/UE – dyrektywa niskonapięciowa LVD obejmująca urządzenia pracujące w zakresie napięć 50…1000 VAC lub 75…1500 VDC,
- 2014/30/UE – dyrektywa kompatybilności elektromagnetycznej (EMC) obejmująca urządzenia zdolne do wytwarzania zaburzeń elektromagnetycznych lub potencjalnie podatne na zakłócenia EMI (czyli… niemal wszystkie urządzenia elektroniczne),
- 2011/65/UE – dyrektywa RoHS 2 obowiązująca obecnie (z wyjątkiem oryginalnego brzmienia załącznika II), a dotycząca ograniczenia stosowania niebezpiecznych substancji (w tym metali ciężkich) w sprzęcie elektrycznym i elektronicznym,
- 2015/863 – tzw. dyrektywa RoHS 3 zmieniająca (rozszerzająca) treść załącznika II poprzez dodanie ograniczeń dot. zawartości ftalanów,
- 2014/53/UE – dyrektywa radiowa (RED) obejmująca urządzenia celowo emitujące i/lub odbierające fale radiowe (poniżej 3 THz).
Warto wspomnieć, że każda z dyrektyw możliwie ściśle określa zarówno zakres wyrobów jej podlegających, jak i zakres wyłączeń. Zdecydowana większość urządzeń elektronicznych (w tym sprzętów codziennego użytku) podlega wymogom dyrektyw LVD, EMC oraz RoHS, zaś w przypadku obecności funkcji radiowych ocena zgodności musi obejmować także zapisy dyrektywy RED (niezależnie od tego, czy urządzenie jest wyposażone w antenę, czy też dostosowane do użycia z anteną zewnętrzną).
Po klasyfikacji wyrobu w zakresie podlegania wymaganiom określonych dyrektyw (rozporządzeń) producent jest zobowiązany dołożyć wszelkich starań, aby przed wprowadzeniem urządzenia do obrotu poprawnie przeprowadzić proces oceny zgodności wyrobu. W efekcie tego etapu wytwórca wystawia deklarację zgodności i nanosi oznakowanie CE na produkt (lub – w niektórych przypadkach – opakowanie bądź dokumentację, o ile umieszczenie czytelnego znaku CE w widocznym miejscu samego wyrobu nie jest możliwe). W procesie oceny zgodności bierze udział nie tylko sam producent – w bardziej zaawansowanych przypadkach konieczny jest udział jednostki notyfikowanej (oznakowanej przez Komisję Europejską unikalnym numerem identyfikacyjnym i uprawnionej do wydawania tzw. certyfikatów zgodności) i laboratoriów akredytowanych, posiadających aktualny certyfikat akredytacji w określonym zakresie wykonywanych badań. Warto jednak pamiętać, że nawet wystawienie certyfikatów przez jednostki notyfikowane i laboratoria (np. EMC) nie jest jeszcze równoznaczne z zielonym światłem do wdrożenia produktu na rynek. Dopiero spełnienie wszystkich wymagań dyrektyw obejmujących swoim zakresem dany wyrób oraz wystawienie przez producenta deklaracji zgodności (poświadczającej, że producent z pełną odpowiedzialnością deklaruje spełnienie wymagań przez dany produkt lub grupę produktów) umożliwia (w większości przypadków) rozpoczęcie sprzedaży urządzeń.
Jednym z wyjątków są właśnie urządzenia medyczne – w ich przypadku konieczne jest przeprowadzenie szeregu dodatkowych procesów, m.in. rejestracji produktu w bazie Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych. W tym artykule nie będziemy jednak zajmować się takimi szczegółami prawnymi, pominiemy także pozostałe istotne aspekty formalne związane z aparaturą medyczną (np. zagadnienia przeprowadzenia tzw. oceny klinicznej, badań klinicznych czy też wdrożenia w przedsiębiorstwie specjalnego systemu kontroli jakości ISO 13485:2016 przeznaczonego dla wyrobów medycznych). Skupimy się natomiast na aspektach związanych z korzystaniem z norm branżowych przez samych konstruktorów, postaramy się także rozwiać kilka pokutujących wśród inżynierów mitów, które skutecznie utrudniają prace przedwdrożeniowe prowadzone przez przedsiębiorstwo i jednocześnie zwiększają ich koszty.
Normy – zmora czy pomocna dłoń?
Podstawowym błędem w rozumieniu istoty norm jest pojmowanie ich przez pryzmat sformalizowanych aktów prawnych, wymuszających na konstruktorach konieczność wykonania dodatkowej, często żmudnej pracy. Mimo że normy zharmonizowane są w rzeczywistości częścią prawa Unii Europejskiej, to jednak według obowiązujących wymagań stosowanie ich podczas opracowywania projektów urządzeń oraz przeprowadzania procesu oceny zgodności jest dobrowolne. Jaki zatem sens ma postępowanie zgodnie z wytycznymi opisanymi w normach? Spełnienie wytycznych normy przez dany produkt pozwala producentowi na zastosowanie tzw. domniemania zgodności z dyrektywami bądź rozporządzeniami. Możliwe jest korzystanie bezpośrednio z zapisów dyrektyw i rozporządzeń unijnych, ale wykorzystanie norm okazuje się w praktyce zdecydowanie łatwiejsze i bardziej przejrzyste.
Nieco o genezie normalizacji
Najistotniejszym faktem z „życia” każdej normy wchodzącej na rynek (bądź aktualizowanej) jest ten, że jej opracowaniem zajmują się wysokiej klasy specjaliści z danej dziedziny, zaś sformułowanie określonych wymagań jest poparte zarówno szeroką wiedzą teoretyczną, jak i realnymi potrzebami rynku oraz, co jeszcze ważniejsze, bogatym doświadczeniem praktycznym. Każda norma stanowi bowiem swego rodzaju zbiór wskazówek, możliwych do bezpośredniego zastosowania w danym projekcie i/lub procesie oceny zgodności. Zaś bieżące aktualizacje oraz wprowadzanie nowych norm, pokrywających kolejne obszary techniki lub zastępujących dawne, wysłużone dokumenty sprawia, że efekty pracy organizacji normalizacyjnych (w Polsce opracowaniem norm zajmuje się Polski Komitet Normalizacyjny, w skrócie PKN) nadążają za rozwojem stanu techniki.
Najważniejszym wnioskiem płynącym z powyższego opisu jest stwierdzenie, że zastosowanie norm już na etapie projektu urządzenia znacznie zwiększa szanse na poprawne, bezproblemowe przejście procesu oceny zgodności. Przekłada się to zresztą na konkretne oszczędności dla przedsiębiorstwa, a doskonałym przykładem będzie tutaj przywołanie dyrektywy EMC. W celu stwierdzenia zgodności z normami zharmonizowanymi dotyczącymi kompatybilności elektromagnetycznej konieczne jest zwykle wykonanie złożonych badań laboratoryjnych i to zarówno w domenie radiowej (komory EMC), jak i w zakresie interferencji przewodzonych (w przypadku obwodów zasilania sieciowego) oraz interfejsów przewodowych (np. wejść pomiarowych czy też portów komunikacyjnych).
Powrót urządzenia z laboratorium w przypadku uzyskania wyniku negatywnego (tj. niespełnienia choćby niewielkiej części wymagań) powoduje konieczność wprowadzenia niezbędnych poprawek w konstrukcji produktu, a następnie… ponownego zlecenia badań. Każda taka iteracja wiąże się ze sporymi kosztami, które można jednak zminimalizować na dwa sposoby:
- Zaprojektowanie urządzenia zgodnie ze sztuką inżynierską i zwrócenie szczególnej uwagi na aspekty kompatybilności elektromagnetycznej na każdym etapie projektu (zastosowanie odpowiedniej architektury obwodów wejścia/wyjścia, użycie filtrów pasywnych i efektywnego ekranowania, właściwe poprowadzenie wewnętrznych wiązek przewodów, uważne zaprojektowanie PCB czy wreszcie zastosowanie zabiegów redukujących poziom emisji i/lub podatności na zakłócenia na etapie tworzenia oprogramowania wbudowanego);
- Wykonanie (znacznie tańszych niż badania kończące się wydaniem raportu z testów laboratoryjnych) badań typu pre-compliance, pozwalających odpowiednio wcześnie namierzyć i zniwelować potencjalne źródła problemów EMI.
Znajomość norm przez konstruktorów pozwala zatem uniknąć wielu problemów – zarówno finansowych (koszty ponownych badań), jak i logistycznych (opóźnienia we wdrożeniu produktu na rynek), a nawet… wizerunkowych.
Właściwa identyfikacja wymagań
Określenie dyrektyw (rozporządzeń), a następnie norm, jakim podlega dany produkt, jest etapem kluczowym dla powodzenia przedsięwzięcia. Okazuje się bowiem, że niektóre dyrektywy wyłączają ze swojego zakresu obowiązywania pewne grupy urządzeń, choć pozornie powinny one podlegać tymże aktom prawnym (przynajmniej w podstawowym zakresie stosowalności). Przykładem są elektryczne (elektroniczne) urządzenia medyczne, które z uwagi na zakres napięć zasilania powinny podlegać dyrektywie LVD, to w istocie są wyłączone spod zakresu dyrektywy niskonapięciowej, podlegając ocenie zgodności opisanej przez Rozporządzenie 2017/745 w sprawie wyrobów medycznych (obowiązujące od 26 maja 2021 i zastępujące całkowicie dawną dyrektywę wyrobów medycznych 93/42/EWG) [1].
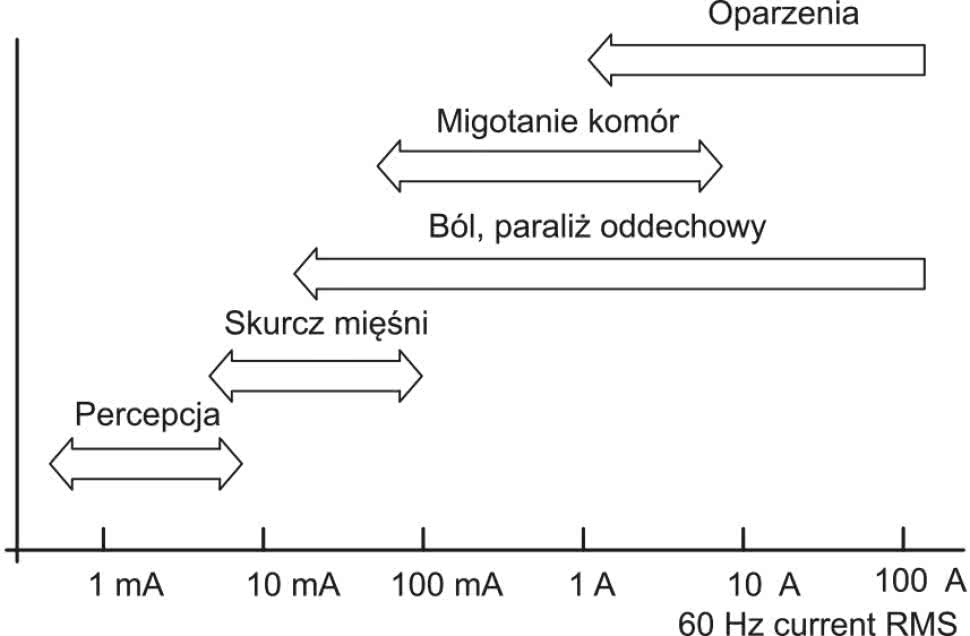
Także normy zharmonizowane dotyczące podstawowego zagadnienia, dla regulacji którego powstała dyrektywa LVD, czyli bezpieczeństwa elektrycznego, są opracowane osobno, specjalnie dla aparatury medycznej i, jak nietrudno się domyślić, operują one znacznie wyższymi wymaganiami niż w przypadku innych rodzajów urządzeń powszechnego użytku (np. sprzętu komputerowego lub RTV). Na rysunku 1 pokazano orientacyjne zakresy wartości prądu przemiennego przepływającego przez ciało człowieka wraz ze wskazaniem wywieranych przez ów prąd skutków biologicznych. W przypadku aparatury medycznej absolutnie niedopuszczalne jest nie tylko wystąpienie umiarkowanie ciężkich lub ciężkich powikłań przepływu prądu przez ciało pacjenta – nawet odczuwanie jakichkolwiek nieprzyjemnych (choć nieszkodliwych z klinicznego punktu widzenia) doznań zdecydowanie nie byłoby pożądane w przypadku urządzeń terapeutycznych i diagnostycznych.
Istotnym aspektem jest też zakres zastosowań danego urządzenia, nierozerwalnie powiązany z potencjalnym ryzykiem stwarzanym przez aparaturę oraz z typem zastosowanej części aplikacyjnej (mającej kontakt z pacjentem). Najwyższa klasa części aplikacyjnej (CF) jest stosowana w odniesieniu do urządzeń mających bezpośredni kontakt z sercem pacjenta, a wymagania dot. prądów upływu są w jej przypadku nad wyraz rygorystyczne [2]. Niższe klasy B oraz BF są stosowane do większości typowych części aplikacyjnych, przy czym klasa BF (lub CF) powinna być wybierana w zastosowaniach do dostarczania energii elektrycznej do ciała pacjenta (np. stymulatory elektryczne) oraz akwizycji sygnałów elektrofizjologicznych (np. tory wejściowe EKG/EEG) – w pozostałych przypadkach możliwe jest zwykle zastosowanie najniższej klasy, tj. części aplikacyjnej typu B.
Warto pamiętać, że niektóre normy mają zastosowanie do danego produktu, nawet jeżeli dana funkcjonalność jest zaledwie jedną z wielu oferowanych przez wyrób. Za przykład złożoności procesu certyfikacji weźmy jedno z podstawowych urządzeń medycznych – monitor pacjenta (kardiomonitor), stosowany do wieloparametrowego monitorowania funkcji życiowych.
Podstawowe normy, które mają zastosowanie do naszego przykładowego urządzenia (jak i zdecydowanej większości innych urządzeń medycznych), to:
- PN-EN 60601-1:2011 (Medyczne urządzenia elektryczne – Część 1: Wymagania ogólne dotyczące bezpieczeństwa podstawowego oraz funkcjonowania zasadniczego);
- PN-EN 60601-1-2:2015-11 (Medyczne urządzenia elektryczne – Część 1-2: Wymagania ogólne dotyczące bezpieczeństwa podstawowego oraz funkcjonowania zasadniczego – Norma uzupełniająca: Zakłócenia elektromagnetyczne – Wymagania i badania);
- PN-EN 60601-1-6:2010 (Medyczne urządzenia elektryczne – Część 1-6: Wymagania ogólne dotyczące bezpieczeństwa podstawowego oraz funkcjonowania zasadniczego – Norma uzupełniająca: Użyteczność);
- PN-EN 60601-1-8:2011 (Medyczne urządzenia elektryczne – Część 1-8: Wymagania ogólne dotyczące bezpieczeństwa podstawowego oraz funkcjonowania zasadniczego – Norma uzupełniająca: Wymagania ogólne, badania i wytyczne dotyczące systemów alarmowych w medycznych urządzeniach elektrycznych i medycznych systemach elektrycznych).
Wszystkie wspomniane normy należą do grupy oznakowanej wspólnym numerem 60601-1(-x) i obejmującej najbardziej fundamentalne wymogi dot. aparatury medycznej. Wymagania te są wspierane przez normy z grupy 60601-2-x, które odnoszą się z kolei do określonych podgrup urządzeń, podzielonych z uwagi na zakres zastosowań i funkcjonalności.
W naszym przykładowym kardiomonitorze oferującym pomiary:
- EKG,
- ciśnienia krwi metodą nieinwazyjną,
- saturacji krwi (SpO2) oraz
- temperatury ciała
będą to najczęściej normy:
- PN-EN 60601-2-27:2014-11 (Medyczne urządzenia elektryczne – Część 2-27: Wymagania szczegółowe dotyczące bezpieczeństwa podstawowego oraz funkcjonowania zasadniczego elektrokardiograficznych urządzeń monitorujących);
- PN-EN IEC 80601-2-30:2019-07 (Medyczne urządzenia elektryczne – Część 2-30: Wymagania szczegółowe dotyczące bezpieczeństwa podstawowego oraz funkcjonowania zasadniczego automatycznych nieinwazyjnych sfigmomanometrów);
- PN-EN ISO 80601-2-61:2019-03 (Medyczne urządzenia elektryczne – Część 2-61: Szczegółowe wymagania dotyczące podstawowego bezpieczeństwa i zasadniczego działania wyposażenia pulsoksymetrów do medycznego stosowania);
- PN-EN ISO 80601-2-56:2017-10 (Medyczne urządzenia elektryczne – Część 2-56: Wymagania szczegółowe dotyczące podstawowego bezpieczeństwa i zasadniczego działania termometrów medycznych do pomiaru temperatury ciała).
Dodatkowo, urządzenie tego rodzaju podlegać będzie normie:
- PN-EN IEC 80601-2-49:2020-03 (Medyczne urządzenia elektryczne – Część 2-49: Wymagania szczegółowe dotyczące bezpieczeństwa podstawowego oraz funkcjonowania zasadniczego urządzeń do wielofunkcyjnego monitorowania pacjenta).
Powyższa lista nie jest oczywiście wyczerpująca, gdyż do wymienionych norm dochodzą jeszcze wymagania dotyczące zarządzania ryzykiem czy też cyklu życia oprogramowania. Nie wspominamy także o sporej liście norm EMC (i ew. radiowych) – do opisanych w nich procedur odnosi się bowiem bezpośrednio „medyczna norma EMC”, czyli 60601-1-2.
Wpływ norm na BOM projektu
Zastosowanie właściwego podejścia do projektowania w oparciu na normach (już od najwcześniejszych etapów opracowywania konstrukcji urządzenia) w praktyce wiąże się nierozerwalnie z umiejętnym wyborem komponentów o znaczeniu kluczowym dla spełnienia konkretnych wymagań. Okazuje się bowiem, że dobór modułów i elementów elektronicznych posiadających własne certyfikaty może znacznie ułatwić opracowanie urządzenia. Zawsze należy mieć jednak na uwadze, że użycie komponentów spełniających określone normy nie jest równoznaczne ze spełnieniem ich przez całe urządzenie. Żelazną zasadą, stosowaną szeroko w analizie ryzyka, jest przyjęcie, że o bezpieczeństwie systemu decyduje jego najsłabszy element.
Zastosowanie właściwych komponentów o znaczeniu krytycznym dla danej aplikacji może jednak znacząco uprościć proces oceny zgodności, gdyż w wielu przypadkach ocena taka opiera się na przeglądzie dokumentacji urządzenia i/lub oględzinach przekazanego do laboratorium egzemplarza testowego.
Kompontenty z „własną certyfikacją”
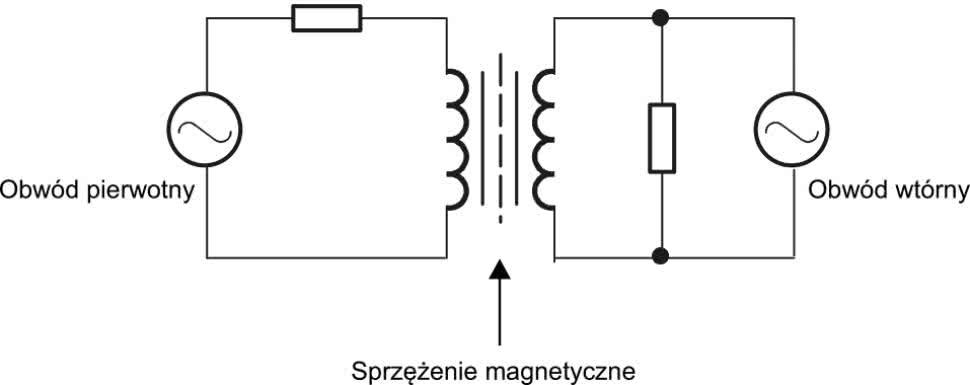
Aby lepiej zilustrować omawiane zagadnienie, posłużymy się przykładem separatorów galwanicznych stosowanych do zapewnienia niezbędnych środków ochrony pacjenta (MOPP) oraz środków ochrony operatora (MOOP), a szeroko opisanych przez przywołaną wcześniej normę 60601-1, która stanowi swoistą biblię wszystkich inżynierów elektroniki medycznej. Zastosowanie separatorów (transoptorów, izolatorów interfejsów szeregowych czy też wzmacniaczy izolacyjnych) umożliwia oddzielenie bezpiecznej części aplikacyjnej (mającej kontakt z ciałem pacjenta – np. front-endu EKG) od nie tak bezpiecznej części urządzenia, kontaktującej się galwanicznie ze światem zewnętrznym. I nie chodzi w tym przypadku tylko o część sieciową zasilacza – także wszelkie porty wejścia/wyjścia (USB, HDMI czy też RS232) mogłyby stanowić istotne zagrożenie dla pacjenta, gdyby poważnej awarii uległ sprzęt współpracujący z rozpatrywanym urządzeniem medycznym, a ścieżka prądu upływu prowadziłaby przez ciało osoby badanej lub poddawanej terapii.
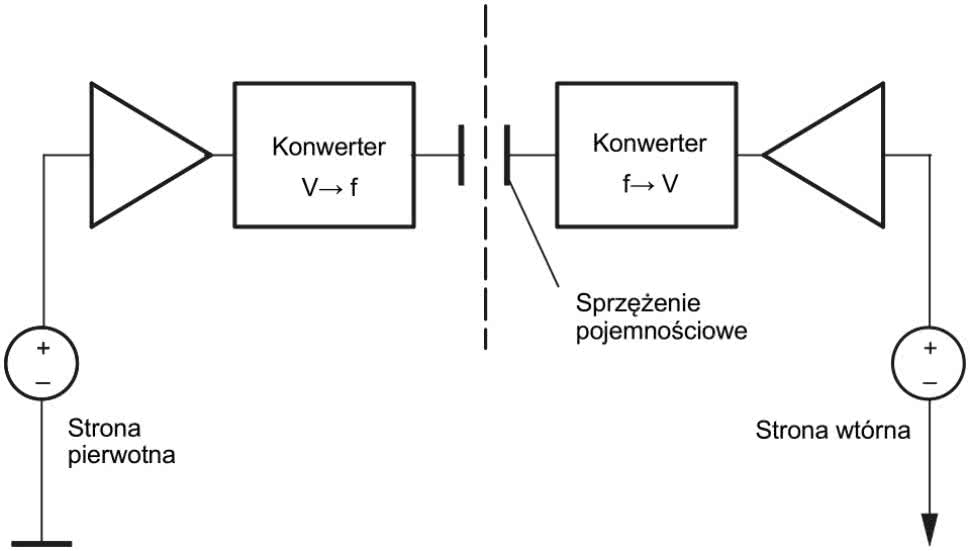
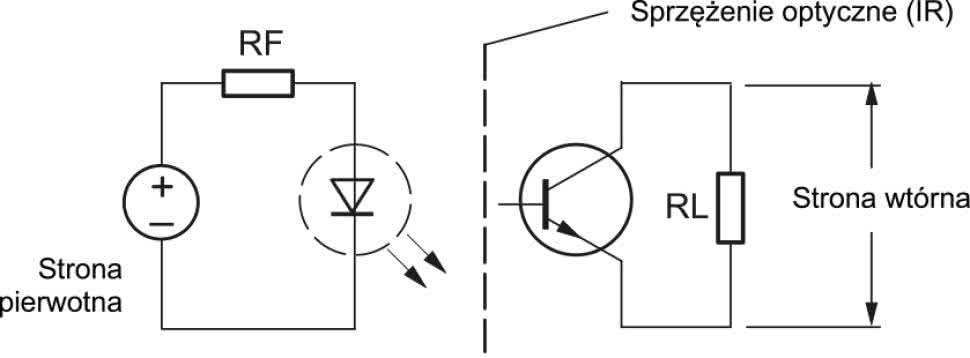
Najczęściej stosowane separatory galwaniczne mają topologię magnetyczną (rysunek 2), pojemnościową (rysunek 3) bądź optyczną (rysunek 4). W przypadku zastosowania separatora scalonego pod uwagę należy wziąć nie tylko wytrzymałość i rezystancję użytego materiału izolacyjnego, ale także uwarunkowania geometryczne, wpływające bezpośrednio na implementację układu w ramach projektu PCB.
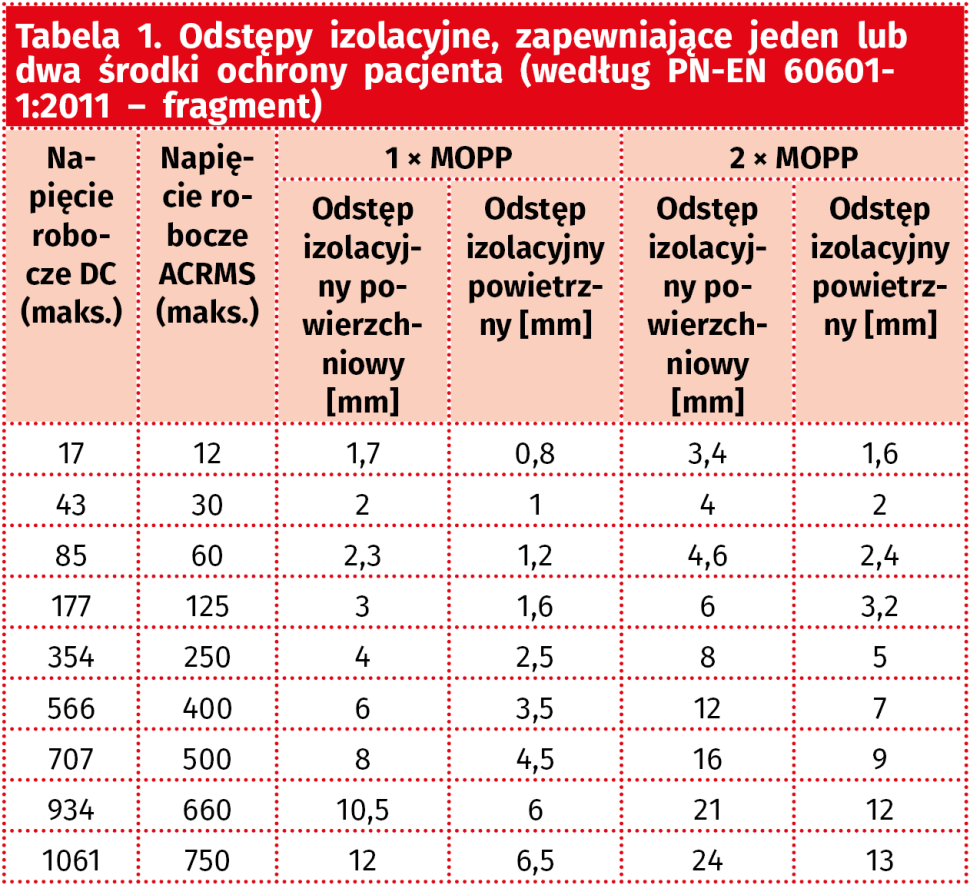
Przykładowo, przywołana wcześniej norma PN-EN 60601-1:2011 definiuje dokładne wymagania dot. minimalnych odstępów izolacyjnych, spełniających kryteria środków ochrony pacjenta (MOPP – tabela 1). Odstępy izolacyjne mogą być definiowane na dwa sposoby: w linii prostej, w powietrzu (clearance) bądź po powierzchni materiału izolacyjnego (z uwzględnieniem najkrótszej możliwej ścieżki pomiędzy dwoma elementami przewodzącymi – creepage) – oba opisane rodzaje odstępów zaprezentowano schematycznie na rysunku 5.

Zastosowanie separatora w urządzeniu zasilanym sieciowo (250 VAC) wymaga zatem wyboru układu, który umożliwi uzyskanie niezbędnych odstępów – w przypadku dwóch środków ochrony pacjenta, wymaganych w celu zabezpieczenia części aplikacyjnych i innych części dostępnych, odstęp powierzchniowy powinien wynosić co najmniej 8 mm, zaś (co także definiuje norma 60601-1) izolacja stała tworząca środek ochrony pacjenta powinna zapewniać wytrzymałość 4 kV (ochrona od części sieciowej zasilacza) lub 3 kV (ochrona od obwodów wtórnych, tj. odseparowanych od części sieciowej co najmniej jednym środkiem ochrony).
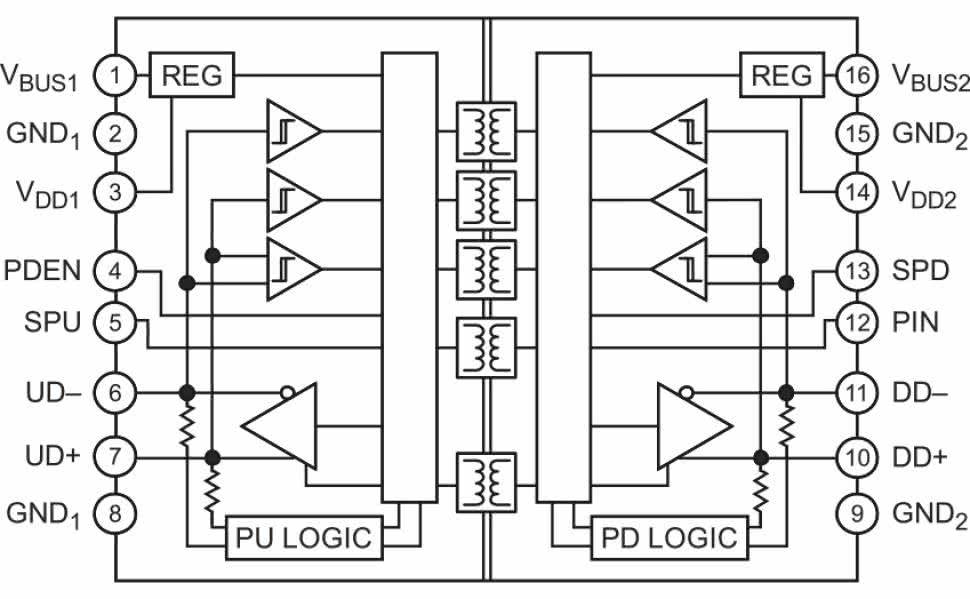
W przypadku konieczności odseparowania pewnej części urządzenia (np. obwodów portu USB) od połączonego bezpośrednio z częścią aplikacyjną (np. front-endem EKG) procesora konieczne okazuje się zatem zastosowanie układu, który zapewni niezbędny poziom bezpieczeństwa elektrycznego. Wymagania takie spełnia izolator ADuM4160 marki Analog Devices (rysunek 6). Wytrzymała izolacja wewnętrzna oraz poszerzona obudowa (rysunek 7) umożliwiają dotrzymanie ww. wymogów, co manifestuje się zapisem w nocie katalogowej układu, wskazującym na spełnienie przezeń wymogów normy IEC 60601-1 dot. izolacji wzmocnionej dla napięcia roboczego równego 250 Vrms.
Warto w tym miejscu podkreślić, że samo użycie izolatora ADuM4160 w omówionym urządzeniu przykładowym nie wystarczy do zapewnienia bezpieczeństwa elektrycznego – konieczne jest bowiem zwrócenie bacznej uwagi na projekt PCB w rejonie izolatora (rozdzielenie pól masy, ścieżek sygnałowych, a także padów samego separatora galwanicznego na odległość >= 8 mm bądź zastosowanie wycięć izolacyjnych w newralgicznych punktach płyty drukowanej), nie wspominając o dopracowaniu całego obwodu zasilania urządzenia oraz wszelkich innych izolacji ochronnych.
Opisany przykład separatora galwanicznego dobrze ilustruje zagadnienie stosowania tzw. podzespołów wysokiej niezawodności, czyli elementów, których potencjalne uszkodzenie mogłoby spowodować nieakceptowalne ryzyko. Paradoksalnie, w takich właśnie przypadkach proces certyfikacji może zostać wydatnie uproszczony dzięki zastosowaniu podzespołów, dla których producent przedstawia dokumentację jednoznacznie stwierdzającą, że jego wyrób spełnia wymagania norm (czyli w praktyce – posiada certyfikat zgodności).
Innym przykładem grupy podzespołów spełniających wymogi określonych norm branżowych będą wszelkiego rodzaju zasilacze sieciowe oraz izolowane przetwornice DC/DC, przeznaczone do aplikacji medycznych. Oferty producentów zasilaczy medycznych – zarówno modułowych (w postaci otwartej, zamkniętej lub przeznaczonej do montażu na PCB), jak i zewnętrznych (wtyczkowych oraz typu desktop) są dziś na tyle szerokie, że zazwyczaj nie stanowi większego problemu dobór odpowiedniego rozwiązania do projektowanej aplikacji.
Parametry zasilaczy medycznych mające bezpośredni wpływ na zachowanie zgodności z normami pokażemy na przykładzie nowoczesnej, niewielkiej przetwornicy modułowej AC/DC z serii TPP 15-J marki Traco Power (fotografia 1). Producent deklaruje prąd upływu [3] poniżej 75 µA, co umożliwia zastosowanie zasilacza w urządzeniach z częścią aplikacyjną typu BF. Norma 60601-1 stawia bowiem dość rygorystyczne wymagania w zakresie dopuszczalnych prądów upływu pacjenta w urządzeniach z częściami aplikacyjnymi typu B i BF, które nie mogą przekraczać 100 µA w stanie normalnym (przy braku awarii) oraz 500 µA w tzw. stanie pojedynczego uszkodzenia. Urządzenie jest wyposażone w zabezpieczenia przeciwzwarciowe, przeciwprzeciążeniowe i przeciwprzepięciowe, zaś jego konstrukcja mechaniczna ułatwia instalację w obudowie docelowego urządzenia (izolująca obudowa modułu, ścianki oddzielające złącza strony wtórnej i sieciowej od otworów montażowych, chłodzenie konwekcyjne bez konieczności stosowania wentylatora).

Komponenty o parametrach ułatwiających spełnienie norm
Nie wszystkie komponenty o kluczowym znaczeniu dla funkcjonowania urządzenia medycznego muszą posiadać określone certyfikaty. Okazuje się jednak, że odpowiedni dobór takich komponentów także może ułatwić certyfikację urządzenia końcowego, a to dzięki parametrom oferowanym (nawet jeśli niepotwierdzonym certyfikatami) przez podzespoły. Podamy dwa przykłady takich rozwiązań wraz z odniesieniem do zapisów konkretnych norm branżowych.
Pomiar temperatury ciała
W przypadku termometrów lekarskich oraz innych systemów pomiaru temperatury ciała pacjentów stosowane mogą być różne normy, z których wymienić warto:
- PN-EN ISO 80601-2-56:2017-10 (Medyczne urządzenia elektryczne – Część 2-56: Wymagania szczegółowe dotyczące podstawowego bezpieczeństwa i zasadniczego działania termometrów medycznych do pomiaru temperatury ciała);
- PN-EN IEC 80601-2-59:2020-01 (Medyczne urządzenia elektryczne – Część 2-59: Wymagania szczegółowe dotyczące bezpieczeństwa podstawowego oraz funkcjonowania zasadniczego termografów do badań przesiewowych temperatury osób w stanie gorączki);
- ASTM E1112 - 00(2018) (Standard Specification for Electronic Thermometer for Intermittent Determination of Patient Temperature).

W przypadku pomiaru stykowego (kontaktowego) zastosowanie na obszarze UE ma pierwsza z wymienionych norm, definiująca podstawową dokładność (zwaną dokładnością laboratoryjną, laboratory accuracy) pomiaru na poziomie ±0,3°C i zakres pomiaru od 34,0 do 43,0°C, zaś w przypadku ASTM E1112 parametry te wynoszą odpowiednio: ±0,1°C oraz 35,8...41,0°C. Zastosowanie czujnika o dokładności ±0,1°C w zakresie (przynajmniej) 35,8...43°C umożliwia zatem spełnienie wymogów obu norm, rozszerzając tym samym potencjalny obszar sprzedaży opartego na sensorze termometru. Przykładem czujnika scalonego, który spełnia ww. wymogi, jest miniaturowy układ TMP117 firmy Texas Instruments (rysunki 8 i 9), oferujący dokładność ±0,1°C w zakresie od –20 do +50°C, bez konieczności dodatkowej kalibracji.

Elektrokardiografia
Wspomniana wcześniej norma PN-EN 60601-2-27:2014-11 opisuje szereg parametrów, które powinny spełniać elektrokardiografy – zarówno w zakresie stopnia wejściowego (front-endu analogowego), jak i przetwarzania sygnałów oraz automatycznej ich analizy. Przykładowe parametry określone wymogami normy to m.in.:
- wejściowy zakres dynamiki: urządzenia EKG powinny obsługiwać sygnały prezentujące offset DC ±300 mV i różnicowy sygnał zmienny na poziomie ±5 mV (wymagany slew rate to 320 mV/s);
- impedancja wejściowa: zalecana wartość wynosi co najmniej 2,5 MΩ;
- szum wejściowy: poziom szumu wprowadzanego przez kabel pacjenta oraz wzmacniacz EKG nie powinien przekraczać 30 µVpp (pomiar przez min. 10 sekund, przy włączonym pasmowozaporowym filtrze częstotliwości sieciowej, jeśli takowy istnieje);
- zakres wzmocnień: urządzenie powinno mieć przynajmniej jedno stałe ustawienie wzmocnienia na poziomie 10 mm/mV ±10%. W większości systemów EKG możliwa jest jednak zmiana wzmocnienia PGA, wtedy test powinien być wykonany dla każdego z nich. Ponadto dryf wzmocnienia nie powinien przekraczać 0,66%/min;
- zalecany minimalny zakres pasma przepustowego wynosi od 0,67 do 40 Hz.
Dostępne na rynku front-endy scalone, przeznaczone do aplikacji elektrokardiograficznych, nie tylko spełniają ww. wymagania, ale oferują parametry wielokrotnie lepsze, dając pewną swobodę projektantowi otoczenia (obwodów peryferyjnych) AFE. Znany front-end EKG marki Texas Instruments – ADS1298(R) oferuje bardzo szeroki zakres dynamiki, znacznie przekraczający parametry wskazane normą (i zależny od szeregu ustawień, w tym wzmocnienia PGA, napięcia zasilania czy też wybranej wartości napięcia odniesienia), a także poziom szumu rzędu 4 µVpp i dokładność oraz stabilność wzmocnienia na poziomie (odpowiednio) ±0,5% i 5 ppm/°C. Więcej szczegółowych informacji na temat front-endów elektrokardiograficznych można znaleźć w trzeciej części „Praktycznych aplikacji scalonych układów AFE” bieżącego wydania „Elektroniki Praktycznej”.
Zastosowanie modułów radiowych w urządzeniach medycznych
Coraz większa liczba urządzeń medycznych jest wyposażona w fabrycznie zainstalowane moduły komunikacji radiowej. Rozwój telemedycyny wymusza na producentach podążanie za potrzebami rynku, co stawia ich przed koniecznością wyboru optymalnego rozwiązania w zakresie komunikacji RF. Podobnie jak w przypadku innych urządzeń elektronicznych, także aparatura medyczna podlega ocenie zgodności w świetle dyrektywy radiowej, o ile tylko jej funkcjonalność obejmuje odbiór i/lub emisję fal radiowych.
Analogicznie do innych obszarów elektroniki, także w tym przypadku można sporo zaoszczędzić na kosztach badań laboratoryjnych, stosując moduły z certyfikatami zgodności dostarczonymi przez producenta. Co więcej, niektórzy wytwórcy modułów RF poczynili ukłon w stronę producentów aparatury medycznej, udostępniając na rynku moduły z obszernym zakresem certyfikacji, obejmującym nie tylko klasyczne normy zharmonizowane dla urządzeń radiowych, ale także część norm medycznych, które mają zastosowanie do modułów RF. Przykładem może być tutaj moduł Wi-Fi NINA-W13 marki u-blox, oferujący certyfikację nie tylko zgodnie z podstawowymi dyrektywami radiowymi dla obszarów Unii Europejskiej, USA, Kanady, Japonii, Tajwanu czy Australii, ale także… medyczną normą EMC 60601-1-2.Więcej informacji na temat modułów Wi-Fi zaprezentowaliśmy w artykule „Moduły Wi-Fi dla IoT” tego wydania EP.
Niezależnie od kwestii typowo elektronicznych nie należy zapominać o wpływie zastosowania komunikacji radiowej na niezawodność urządzenia i jego funkcjonowanie zasadnicze. Ważnymi zagadnieniami do opracowania są m.in. ochrona danych osobowych/medycznych, zabezpieczenia teleinformatyczne (szyfrowanie, uwierzytelnianie), itd.
Jak bezboleśnie przejść certyfikację wyrobu?
Uwzględnienie wymogów formalnych już na początku pracy nad projektem znakomicie ułatwia i przyspiesza proces wdrożenia rynkowego gotowego wyrobu. Zapewne niejeden spośród Czytelników zada sobie jednak w tym momencie pytanie – co zrobić, jeżeli produkt jest już w trakcie opracowania i nie ma możliwości zaprojektowania go od początku z myślą o normach branżowych? Zbierzmy zatem garść praktycznych porad dotyczących projektowania urządzeń elektronicznych – w tym celu posłużymy się znów przykładami z branży aparatury medycznej.
- Określenie dyrektyw, rozporządzeń i konkretnych norm zharmonizowanych, obejmujących swoim zakresem opracowywany produkt, praktycznie nigdy nie oznacza konieczności spełnienia wszystkich ich zapisów. Należy zwrócić baczną uwagę na te podrozdziały i punkty, które rzeczywiście dotyczą danego wyrobu.
Przykład – urządzenia medyczne mogą podlegać wymaganiom dotyczącym ochrony przed zagrożeniem wywołania zapłonu łatwopalnych substancji (mieszanin anestetycznych lub palnych środków do dezynfekcji, lub czyszczenia skóry), stosowanych m.in. na blokach operacyjnych. Wymagania te implikują ograniczenia dopuszczalnej energii gromadzonej w wewnętrznych komponentach pasywnych (kondensatory i elementy indukcyjne) oraz szereg dodatkowych obostrzeń. Ta część wymagań nie ma jednak zastosowania do urządzeń medycznych, które w żadnym przypadku nie będą pracowały w takim otoczeniu, co opisuje punkt G1.1 normy 60601-1; - Część testów można wykonać samodzielnie w ramach prototypowni – znacznym ułatwieniem jest dostęp do aparatury pomiarowej i wyposażenia, które umożliwi przeprowadzenie określonych typów badań, opisanych w normie. Na rynku polskim istnieją firmy, które w ramach kompleksowej działalności badawczo-rozwojowej są w stanie nie tylko opracować urządzenie koncepcyjnie i technicznie (do etapu prototypu, a nawet produkcji), ale także wykonać szereg badań we własnym zakresie. Często nawiązanie współpracy z tego typu przedsiębiorstwem daje wymierne korzyści zlecającemu, który nie musi inwestować w aparaturę pomiarową (często bardzo kosztowną). Najprostsze testy nie wymagają natomiast dostępu do skomplikowanych przyrządów laboratoryjnych i można je wykonać za pomocą prostych środków, dostępnych niemal od ręki.
Przykład – norma 60601-1 definiuje szereg testów dot. wytrzymałości mechanicznej urządzenia. Przyrządy ręczne należy testować poprzez upuszczenie ich na twarde podłoże z wysokości 1 metra, z kolei większe urządzenia przenośne, zależnie od masy, z wysokości od 2 do 5 cm. Testy takie można z powodzeniem wykonać bez ponoszenia większych kosztów, gdyż wystarczy do tego celu... 5-centymetrowa płyta z twardego drewna, umieszczona na podłodze betonowej (lub podobnej); - Badania typu pre-compliance – część testów warto wykonać w specjalistycznym laboratorium, które nie tylko udostępni wyniki wstępnych pomiarów, ale także posłuży wsparciem merytorycznym. Taka sytuacja ma miejsce przede wszystkim w przypadku badań EMC, gdyż stosunkowo niewiele firm projektowych i produkcyjnych może pozwolić sobie na wybudowanie i wyposażenie własnej komory EMC.
Przykład – zamówienie badań pre-compliance w laboratorium EMC pozwala namierzyć i scharakteryzować źródła potencjalnych zakłóceń, które mogłyby spowodować uzyskanie negatywnego wyniku pełnych testów certyfikacyjnych. Ma to znaczenie zwłaszcza w przypadku bardziej złożonych urządzeń cyfrowych z szybkimi procesorami bądź układami FPGA (emisja zakłóceń RF od obwodów komunikacyjnych i taktowania), choć rzecz jasna testy EMC dotyczą w równym stopniu wszystkich urządzeń elektronicznych; - Na zaawansowanych etapach pracy nad projektem (np. po uruchomieniu prototypu) warto zwrócić się o pomoc do specjalisty, który wykona preaudyt opracowywanego urządzenia, zwracając uwagę na istniejące lub potencjalne źródła problemów z zachowaniem zgodności z normami. Doradzi on także możliwe rozwiązania, które mają na celu zwiększenie prawdopodobieństwa pomyślnego przejścia wszystkich testów laboratoryjnych już w ich pierwszej iteracji.
Przykład – z doświadczenia własnego autora wynika, jak wiele mankamentów można wskazać obserwując jedynie samą obudowę urządzenia, sposób montażu wewnętrznego okablowania i złączy. Prostym przykładem jest punkt 8.6.2 normy 60601-1, zakazujący stosowania zacisków uziemienia ochronnego do mechanicznego mocowania elementów niezwiązanych z uziemieniem jako takim – we wspomnianym preaudycie autor znalazł we wnętrzu poddawanego oględzinom urządzenia… zacisk uziemienia, pełniący jednocześnie funkcję elementu przytwierdzającego obudowę sterownika do konstrukcji urządzenia. - Oprogramowanie (także wbudowane) jest częścią wyrobu medycznego i jako takie podlega certyfikacji, co więcej – z zastosowaniem dodatkowych norm, dotyczących m.in. cyklu życia oprogramowania. Użycie odpowiednich procedur już na początku prac software'owych ułatwia późniejsze opracowanie dokumentacji, niezbędnej m.in. do wdrożenia systemu kontroli jakości (w przypadku wyrobów medycznych jest to system ISO 13485:2016) oraz zarządzania ryzykiem wyrobów medycznych (PN-EN ISO 14971:2020-05).
Przykład – zastosowanie oprogramowania gotowego lub wytworzonego przez osoby trzecie wymaga od producenta wyrobu medycznego ustalenia dokładnych wymagań dla takiego oprogramowania, ponadto – co opisuje kolejny punkt normy 60601-1 (H3.4), konieczne jest właściwe zarządzanie procesami integracji i testów, w tym także uwzględniających przypadki najbardziej niekorzystne. Zagadnienia te są szczególnie ważne już na początku tworzenia oprogramowania – w tym podczas wyboru systemu operacyjnego (np. RTOS) czy też wyboru zewnętrznych bibliotek programistycznych.
Podsumowanie
Przedstawione w artykule informacje mają za zadanie nakreślić ogólny obraz procesu projektowania z uwzględnieniem norm branżowych już od początkowych etapów pracy – wyboru funkcjonalności, architektury urządzenia i wreszcie – listy kluczowych komponentów. Każda branża rządzi się swoimi prawami i jako taka wymagałaby osobnego cyklu artykułów, by choć częściowo przybliżyć tę złożoną tematykę. Opisane tutaj zagadnienia, związane z wymogami stawianymi przed współczesną aparaturą medyczną, doskonale obrazują, jak wiele można zyskać, stosując odpowiednie podejście do projektu i do samych norm, które – wbrew pozorom mają za zadanie pomóc konstruktorom, a nie utrudnić im życie.
Zaprezentowane w niniejszym artykule informacje poglądowe należy traktować jako ogólne wskazówki i nakreślenie tematyki normalizacji w procesie projektowania oraz wdrażania urządzeń. Autor oraz Redakcja nie biorą odpowiedzialności za skutki zastosowania tych informacji w realnych aplikacjach (w tym w urządzeniach medycznych). Konstruktorzy producenci są każdorazowo zobowiązani do samodzielnej weryfikacji informacji z treścią odpowiednich norm, aktów prawnych oraz dokumentacji opisanych komponentów.
inż. Przemysław Musz, EP
przemyslaw.musz@ep.com.pl
- W rzeczywistości nie wszystkie urządzenia medyczne są objęte rozporządzeniem 2017/745. Wyjątkiem są aktywne implanty (dyrektywa 90/385/EWG) oraz aparatura i wyposażenie do diagnostyki in vitro (rozporządzenie 2017/746, które wejdzie w życie 26.05.2022 i zastąpi dyrektywę 98/79/WE).
- Litera F w oznaczeniu oznacza tzw. „pływającą” (izolowaną) część aplikacyjną – oznacza to, że blok urządzenia mający kontakt z ciałem pacjenta jest skutecznie odseparowany od reszty urządzenia w celu ograniczenia prądu upływu pacjenta do bezpiecznej wartości, określonej normą 60601-1.
- W przypadku omawianego zasilacza nie zdefiniowano prądu upływu uziomowego, gdyż moduł pracuje w II klasie ochronności.












